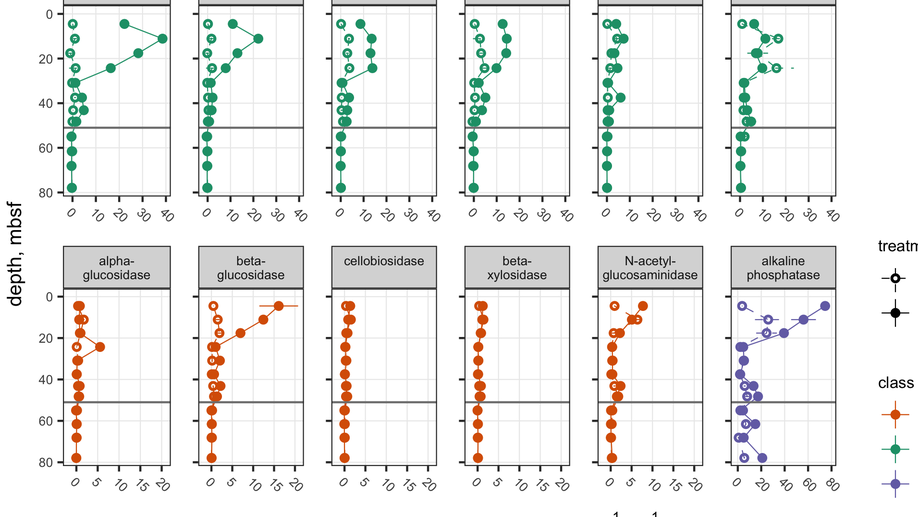
Heterotrophic microorganisms in marine sediments produce extracellular enzymes to hydrolyze organic macromolecules, so their products can be transported inside the cell and used for energy and growth. Therefore, extracellular enzymes may mediate the fate of organic carbon in sediments. The Baltic Sea Basin is a primarily depositional environment with high potential for organic matter preservation. The potential activities of multiple organic carbon-degrading enzymes were measured in samples obtained by the International Ocean Discovery Program Expedition 347 from the Little Belt Strait, Denmark, core M0059C. Potential maximum hydrolysis rates (Vmax) were measured at depths down to 77.9mbsf for the following enzymes: alkaline phosphatase, β-D-xylosidase, β-D-cellobiohydrolase, N-acetyl-β-D-glucosaminidase, β-glucosidase, α-glucosidase, leucyl aminopeptidase, arginyl aminopeptidase, prolyl aminopeptidase, gingipain, and clostripain. Extracellular peptidase activities were detectable at depths shallower than 54.95mbsf, and alkaline phosphatase activity was detectable throughout the core, albeit against a relatively high activity in autoclaved sediments. β-glucosidase activities were detected above 30mbsf; however, activities of other glycosyl hydrolases (β-xylosidase, β-cellobiohydrolase, N-acetyl-β-glucosaminidase, and α-glucosidase) were generally indistinguishable from zero at all depths. These extracellular enzymes appear to be extremely stable: Among all enzymes, a median of 51.3% of enzyme activity was retained after autoclaving for an hour. We show that enzyme turnover times scale with the inverse of community metabolic rates, such that enzyme lifetimes in subsurface sediments, in which metabolic rates are very slow, are likely to be extraordinarily long. A back-of-the-envelope calculation suggests enzyme lifetimes are, at minimum, on the order of 230days, and may be substantially longer. These results lend empirical support to the hypothesis that a population of subsurface microbes persist by using extracellular enzymes to slowly metabolize old, highly degraded organic carbon.
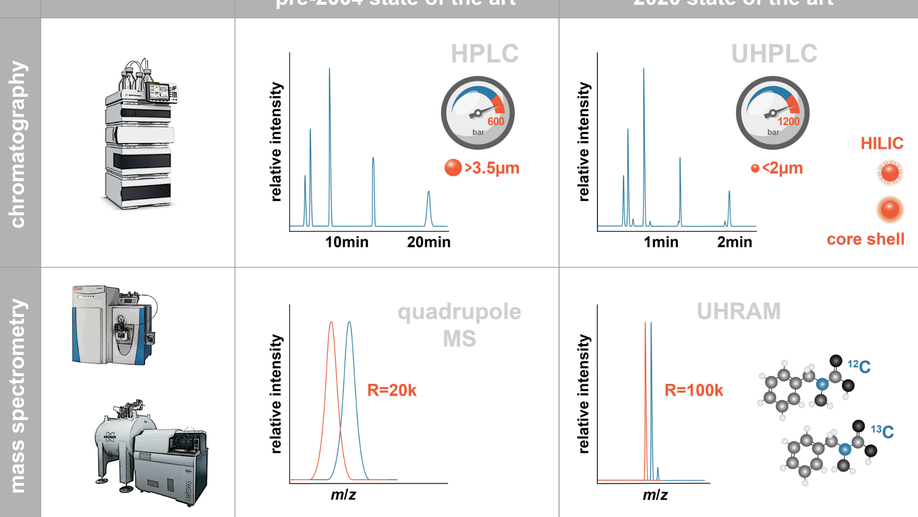
Advances in sampling tools, analytical methods, and data handling capabilities have been fundamental to the growth of marine organic biogeochemistry over the past four decades. There has always been a strong feedback between analytical advances and scientific advances. However, whereas advances in analytical technology were often the driving force that made possible progress in elucidating the sources and fate of organic matter in the ocean in the first decades of marine organic biogeochemistry, today process-based scientific questions should drive analytical developments. Several paradigm shifts and challenges for the future are related to the intersection between analytical progress and scientific evolution. Untargeted “molecular headhunting” for its own sake is now being subsumed into process-driven targeted investigations that ask new questions and thus require new analytical capabilities. However, there are still major gaps in characterizing the chemical composition and biochemical behavior of macromolecules, as well as in generating reference standards for relevant types of organic matter. Field-based measurements are now routinely complemented by controlled laboratory experiments and in situ rate measurements of key biogeochemical processes. And finally, the multidisciplinary investigations that are becoming more common generate large and diverse datasets, requiring innovative computational tools to integrate often disparate data sets, including better global coverage and mapping. Here, we compile examples of developments in analytical methods that have enabled transformative scientific advances since 2004, and we project some challenges and opportunities in the near future. We believe that addressing these challenges and capitalizing on these opportunities will ensure continued progress in understanding the cycling of organic carbon in the ocean.
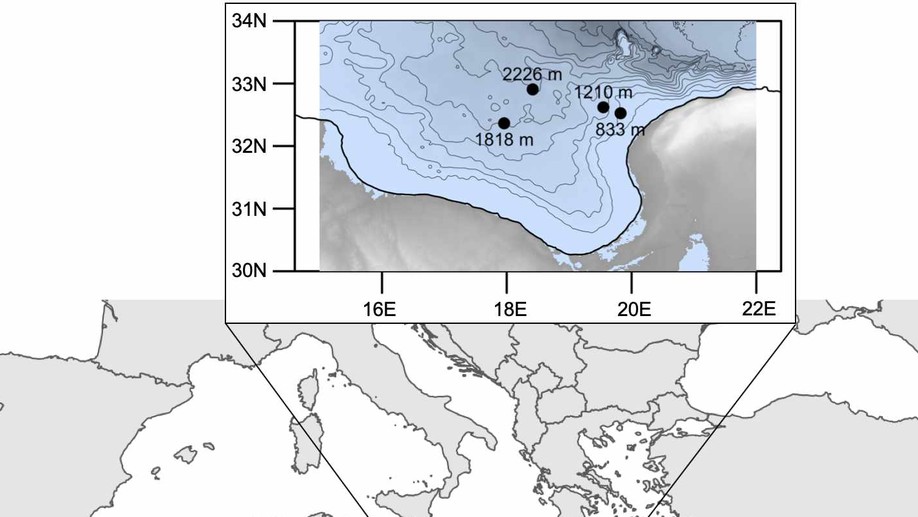
Deep-sea sediments are populated by diverse microbial communities that derive their nutritional requirements from the degradation of organic matter. Extracellular hydrolytic enzymes play a key role in the survival of microbes by enabling them to access and degrade complex organic compounds that are found in seafloor sediments. Despite their importance, extracellular enzymatic activity is poorly characterized at water depths greater than a few hundred meters where physical properties, such as pressure and temperature, create a unique environment for influencing enzyme behavior. Here, we investigated microbial communities and enzyme activities in surface sediment collected at four sampling stations in the central Mediterranean Sea at water depths ranging from 800 to 2200 m. Fluorometric assays revealed that extracellular hydrolytic activity varied according to substrate type and water depth which suggests that the distributions of these enzymes within this basin are not homogenous. Furthermore, enzyme activities indicated substantial demand for phosphomonoesters and proteins, with measurable but much lower demand for polysaccharides. Barcoded amplicon sequencing of bacterial and archaeal SSU genes revealed that microbial communities varied across sampling stations and some groups displayed water-depth related trends. Our results demonstrate that heterotrophic capabilities of microbes in deep-sea Mediterranean sediments can differ substantially even within the same region.
Gene annotation has traditionally required direct comparison of DNA sequences between an unknown gene and a database of known ones using string comparison methods. However, these methods do not provide useful information when a gene does not have a close match in the database. In addition, each comparison can be costly when the database is large since it requires alignments and a series of string comparisons. In this work we propose a novel approach: using recurrent neural networks to embed DNA or amino-acid sequences in a low-dimensional space in which distances correlate with functional similarity. This embedding space overcomes both shortcomings of the method of aligning sequences and comparing homology. First, it allows us to obtain information about genes which do not have exact matches by measuring their similarity to other ones in the database. If our database is labeled this can provide labels for a query gene as is done in traditional methods. However, even if the database is unlabeled it allows us to find clusters and infer some characteristics of the gene population. In addition, each comparison is much faster than traditional methods since the distance metric is reduced to the Euclidean distance, and thus efficient approximate nearest neighbor algorithms can be used to find the best match. We present results showing the advantage of our algorithm. More specifically we show how our embedding can be useful for both classification tasks when our labels are known, and clustering tasks where our sequences belong to classes which have not been seen before.
A recent paper by Martiny argues that “high proportions” of bacteria in diverse Earth environments have been cultured. Here we reanalyze a portion of the data in that paper, and argue that the conclusion is based on several technical errors, most notably a calculation of sequence similarity that does not account for sequence gaps, and the reliance on 16S rRNA gene amplicons that are known to be biased towards cultured organisms. We further argue that the paper is also based on a conceptual error: namely, that sequence similarity cannot be used to infer “culturability” because one cannot infer physiology from 16S rRNA gene sequences. Combined with other recent, more reliable studies, the evidence supports the conclusion that most bacterial and archaeal taxa remain uncultured.
Anoxic subsurface sediments contain communities of heterotrophic microorganisms that metabolize organic carbon at extraordinarily slow rates. In order to assess the mechanisms by which subsurface microorganisms access detrital sedimentary organic matter, we measured kinetics of a range of extracellular peptidases in anoxic sediments of the White Oak River estuary, NC. Nine distinct peptidase substrates were enzymatically hydrolyzed at all depths. Potential peptidase activities (Vmax) decreased with increasing sediment depth, although Vmax expressed on a per cell basis was approximately the same at all depths. Half-saturation constants (Km) decreased with depth, indicating peptidases that functioned more efficiently at low substrate concentrations. Potential activities of extracellular peptidases acting on molecules that are enriched in degraded organic matter (D-phenylalanine and L-ornithine) increased relative to enzymes that act on L-phenylalanine, further suggesting microbial community adaptation to access degraded organic matter. Nineteen classes of predicted, exported peptidases were identified in genomic data from the same site, of which genes for class C25 (gingipain-like) peptidases represented more than 40% at each depth. Methionine aminopeptidases, zinc carboxypeptidases, and class S24-like peptidases, which are involved in single-stranded DNA repair, were also abundant. These results suggest a subsurface heterotrophic microbial community that primarily accesses low-quality detrital organic matter via a diverse suite of well-adapted extracellular enzymes.
Proteins constitute a particularly bioavailable subset of organic carbon and nitrogen in aquatic environments but must be hydrolyzed by extracellular enzymes prior to being metabolized by microorganisms. Activities of extracellular peptidases (protein-degrading enzymes) have frequently been assayed in freshwater systems, but such studies have been limited to substrates for a single enzyme [leucyl aminopeptidase (Leu-AP)] out of more than 300 biochemically recognized peptidases. Here, we report kinetic measurements of extracellular hydrolysis of five substrates in 28 freshwater bodies in the Delaware Water Gap National Recreation Area in the Pocono Mountains (PA, United States) and near Knoxville (TN, United States), between 2013 and 2016. The assays putatively test for four aminopeptidases (arginyl aminopeptidase, glyclyl aminopeptidase, Leu-AP, and pyroglutamyl aminopeptidase), which cleave N-terminal amino acids from proteins, and trypsin, an endopeptidase, which cleaves proteins mid-chain. Aminopeptidase and the trypsin-like activity were observed in all water bodies, indicating that a diverse set of peptidases is typical in freshwater. However, ratios of peptidase activities were variable among sites: aminopeptidases dominated at some sites and trypsin-like activity at others. At a given site, the ratios remained fairly consistent over time, indicating that they are driven by ecological factors. Studies in which only Leu-AP activity is measured may underestimate the total peptidolytic capacity of an environment, due to the variable contribution of endopeptidases.
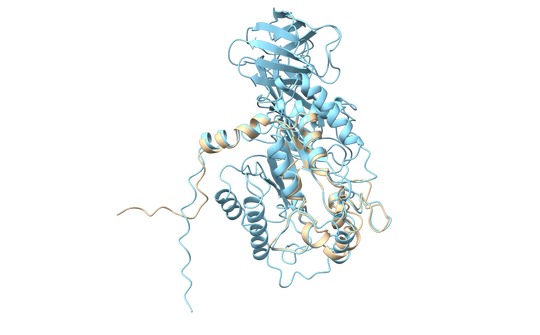
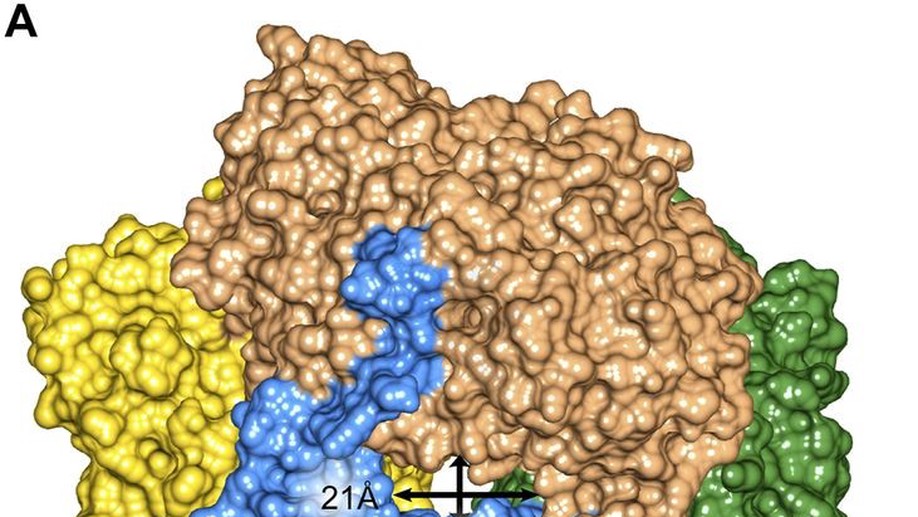
Marine sediments host a large population of diverse, heterotrophic, uncultured microorganisms with unknown physiologies that control carbon flow through organic matter decomposition. Recently, single-cell genomics uncovered new key players in these processes, such as the miscellaneous crenarchaeotal group. These widespread archaea encode putative intra- and extracellular proteases for the degradation of detrital proteins present in sediments. Here, we show that one of these enzymes is a self-compartmentalizing tetrameric aminopeptidase with a preference for cysteine and hydrophobic residues at the N terminus of the hydrolyzed peptide. The ability to perform detailed characterizations of enzymes from native subsurface microorganisms, without requiring that those organisms first be grown in pure culture, holds great promise for understanding key carbon transformations in the environment as well as identifying new enzymes for biomedical and biotechnological applications.
To describe a microbe’s physiology, including its metabolism, environmental roles, and growth characteristics, it must be grown in a laboratory culture. Unfortunately, many phylogenetically novel groups have never been cultured, so their physiologies have only been inferred from genomics and environmental characteristics. Although the diversity, or number of different taxonomic groups, of uncultured clades has been studied well, their global abundances, or numbers of cells in any given environment, have not been assessed. We quantified the degree of similarity of 16S rRNA gene sequences from diverse environments in publicly available metagenome and metatranscriptome databases, which we show have far less of the culture bias present in primer-amplified 16S rRNA gene surveys, to those of their nearest cultured relatives. Whether normalized to scaffold read depths or not, the highest abundances of metagenomic 16S rRNA gene sequences belong to phylogenetically novel uncultured groups in seawater, freshwater, terrestrial subsurface, soil, hypersaline environments, marine sediment, hot springs, hydrothermal vents, nonhuman hosts, snow, and bioreactors (22% to 87% uncultured genera to classes and 0% to 64% uncultured phyla). The exceptions were human and human-associated environments, which were dominated by cultured genera (45% to 97%). We estimate that uncultured genera and phyla could comprise 7.3 × 1029 (81%) and 2.2 × 1029 (25%) of microbial cells, respectively. Uncultured phyla were overrepresented in metatranscriptomes relative to metagenomes (46% to 84% of sequences in a given environment), suggesting that they are viable. Therefore, uncultured microbes, often from deeply phylogenetically divergent groups, dominate nonhuman environments on Earth, and their undiscovered physiologies may matter for Earth systems.
The assembly of single-amplified genomes (SAGs) and metagenome-assembled genomes (MAGs) has led to a surge in genome-based discoveries of members affiliated with Archaea and Bacteria, bringing with it a need to develop guidelines for nomenclature of uncultivated microorganisms. The International Code of Nomenclature of Prokaryotes (ICNP) only recognizes cultures as ‘type material’, thereby preventing the naming of uncultivated organisms. In this Consensus Statement, we propose two potential paths to solve this nomenclatural conundrum. One option is the adoption of previously proposed modifications to the ICNP to recognize DNA sequences as acceptable type material; the other option creates a nomenclatural code for uncultivated Archaea and Bacteria that could eventually be merged with the ICNP in the future. Regardless of the path taken, we believe that action is needed now within the scientific community to develop consistent rules for nomenclature of uncultivated taxa in order to provide clarity and stability, and to effectively communicate microbial diversity.
Heterotrophic microorganisms in marine sediments produce extracellular enzymes to hydrolyze organic macromolecules, so their products can be transported inside the cell and used for energy and growth. Therefore, extracellular enzymes may mediate the fate of organic carbon in sediments. The Baltic Sea Basin is a primarily depositional environment with high potential for organic matter preservation. The potential activities of multiple organic carbon-degrading enzymes were measured in samples obtained by the International Ocean Discovery Program Expedition 347 from the Little Belt Strait, Denmark, core M0059C. Potential maximum hydrolysis rates (Vmax) were measured at depths down to 77.9mbsf for the following enzymes: alkaline phosphatase, β-D-xylosidase, β-D-cellobiohydrolase, N-acetyl-β-D-glucosaminidase, β-glucosidase, α-glucosidase, leucyl aminopeptidase, arginyl aminopeptidase, prolyl aminopeptidase, gingipain, and clostripain. Extracellular peptidase activities were detectable at depths shallower than 54.95mbsf, and alkaline phosphatase activity was detectable throughout the core, albeit against a relatively high activity in autoclaved sediments. β-glucosidase activities were detected above 30mbsf; however, activities of other glycosyl hydrolases (β-xylosidase, β-cellobiohydrolase, N-acetyl-β-glucosaminidase, and α-glucosidase) were generally indistinguishable from zero at all depths. These extracellular enzymes appear to be extremely stable: Among all enzymes, a median of 51.3% of enzyme activity was retained after autoclaving for an hour. We show that enzyme turnover times scale with the inverse of community metabolic rates, such that enzyme lifetimes in subsurface sediments, in which metabolic rates are very slow, are likely to be extraordinarily long. A back-of-the-envelope calculation suggests enzyme lifetimes are, at minimum, on the order of 230days, and may be substantially longer. These results lend empirical support to the hypothesis that a population of subsurface microbes persist by using extracellular enzymes to slowly metabolize old, highly degraded organic carbon.
Advances in sampling tools, analytical methods, and data handling capabilities have been fundamental to the growth of marine organic biogeochemistry over the past four decades. There has always been a strong feedback between analytical advances and scientific advances. However, whereas advances in analytical technology were often the driving force that made possible progress in elucidating the sources and fate of organic matter in the ocean in the first decades of marine organic biogeochemistry, today process-based scientific questions should drive analytical developments. Several paradigm shifts and challenges for the future are related to the intersection between analytical progress and scientific evolution. Untargeted “molecular headhunting” for its own sake is now being subsumed into process-driven targeted investigations that ask new questions and thus require new analytical capabilities. However, there are still major gaps in characterizing the chemical composition and biochemical behavior of macromolecules, as well as in generating reference standards for relevant types of organic matter. Field-based measurements are now routinely complemented by controlled laboratory experiments and in situ rate measurements of key biogeochemical processes. And finally, the multidisciplinary investigations that are becoming more common generate large and diverse datasets, requiring innovative computational tools to integrate often disparate data sets, including better global coverage and mapping. Here, we compile examples of developments in analytical methods that have enabled transformative scientific advances since 2004, and we project some challenges and opportunities in the near future. We believe that addressing these challenges and capitalizing on these opportunities will ensure continued progress in understanding the cycling of organic carbon in the ocean.
Deep-sea sediments are populated by diverse microbial communities that derive their nutritional requirements from the degradation of organic matter. Extracellular hydrolytic enzymes play a key role in the survival of microbes by enabling them to access and degrade complex organic compounds that are found in seafloor sediments. Despite their importance, extracellular enzymatic activity is poorly characterized at water depths greater than a few hundred meters where physical properties, such as pressure and temperature, create a unique environment for influencing enzyme behavior. Here, we investigated microbial communities and enzyme activities in surface sediment collected at four sampling stations in the central Mediterranean Sea at water depths ranging from 800 to 2200 m. Fluorometric assays revealed that extracellular hydrolytic activity varied according to substrate type and water depth which suggests that the distributions of these enzymes within this basin are not homogenous. Furthermore, enzyme activities indicated substantial demand for phosphomonoesters and proteins, with measurable but much lower demand for polysaccharides. Barcoded amplicon sequencing of bacterial and archaeal SSU genes revealed that microbial communities varied across sampling stations and some groups displayed water-depth related trends. Our results demonstrate that heterotrophic capabilities of microbes in deep-sea Mediterranean sediments can differ substantially even within the same region.
Organic carbon in marine sediments is a critical component of the global carbon cycle, and its degradation influences a wide range of phenomena, including the magnitude of carbon sequestration over geologic timescales, the recycling of inorganic carbon and nutrients, the dissolution and precipitation of carbonates, the production of methane and the nature of the seafloor biosphere. Although much has been learned about the factors that promote and hinder rates of organic carbon degradation in natural systems, the controls on the distribution of organic carbon in modern and ancient sediments are still not fully understood. In this review, we summarize how recent findings are changing entrenched perspectives on organic matter degradation in marine sediments: a shift from a structurally-based chemical reactivity viewpoint towards an emerging acceptance of the role of the ecosystem in organic matter degradation rates. That is, organic carbon has a range of reactivities determined by not only the nature of the organic compounds, but by the biological, geochemical, and physical attributes of its environment. This shift in mindset has gradually come about due to a greater diversity of sample sites, the molecular revolution in biology, discoveries concerning the extent and limits of life, advances in quantitative modeling, investigations of ocean carbon cycling under a variety of extreme paleo-conditions (e.g. greenhouse environments, euxinic/anoxic oceans), the application of novel analytical techniques and interdisciplinary efforts. Adopting this view across scientific disciplines will enable additional progress in understanding how marine sediments influence the global carbon cycle.
This chapter is in Deep Carbon: Past to Present, Beth N. Orcutt, Isabelle Daniel, and Rajdeep Gupta, eds, to be published by Cambrdige University Press in October 2019.
Gene annotation has traditionally required direct comparison of DNA sequences between an unknown gene and a database of known ones using string comparison methods. However, these methods do not provide useful information when a gene does not have a close match in the database. In addition, each comparison can be costly when the database is large since it requires alignments and a series of string comparisons. In this work we propose a novel approach: using recurrent neural networks to embed DNA or amino-acid sequences in a low-dimensional space in which distances correlate with functional similarity. This embedding space overcomes both shortcomings of the method of aligning sequences and comparing homology. First, it allows us to obtain information about genes which do not have exact matches by measuring their similarity to other ones in the database. If our database is labeled this can provide labels for a query gene as is done in traditional methods. However, even if the database is unlabeled it allows us to find clusters and infer some characteristics of the gene population. In addition, each comparison is much faster than traditional methods since the distance metric is reduced to the Euclidean distance, and thus efficient approximate nearest neighbor algorithms can be used to find the best match. We present results showing the advantage of our algorithm. More specifically we show how our embedding can be useful for both classification tasks when our labels are known, and clustering tasks where our sequences belong to classes which have not been seen before.
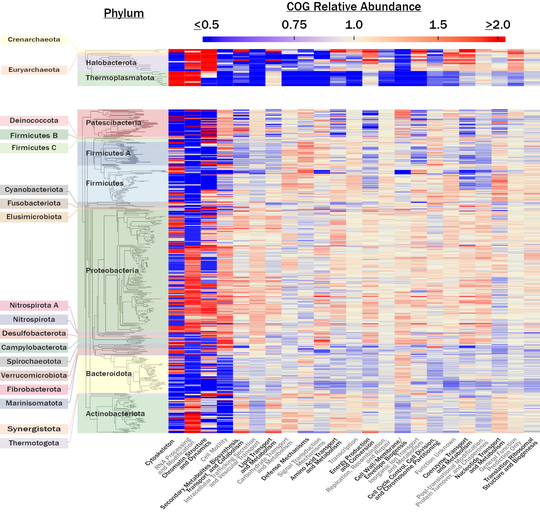
Widely used microbial taxonomies, such as the NCBI taxonomy, are based on a combination of sequence homology among conserved genes and historically accepted taxonomies, which were developed based on observable traits such as morphology and physiology. A recently proposed alternative taxonomy database, the Genome Taxonomy Database (GTDB), incorporates only sequence homology of conserved genes and attempts to partition taxonomic ranks such that each rank implies the same amount of evolutionary distance, regardless of its position on the phylogenetic tree. This provides the first opportunity to completely separate taxonomy from traits and therefore to quantify how taxonomic rank corresponds to traits across the microbial tree of life. We quantified the relative abundances of clusters of orthologous group functional categories (COG-FCs) as a proxy for traits within the lineages of 13,735 cultured and uncultured microbial lineages from a custom-curated genome database. On average, 41.4% of the variation in COG-FC relative abundance is explained by taxonomic rank, with domain, phylum, class, order, family, and genus explaining, on average, 3.2%, 14.6%, 4.1%, 9.2%, 4.8%, and 5.5% of the variance, respectively (P < 0.001 for all). To our knowledge, this is the first work to quantify the variance in metabolic potential contributed by individual taxonomic ranks. A qualitative comparison between the COG-FC relative abundances and genus-level phylogenies, generated from published concatenated protein sequence alignments, further supports the idea that metabolic potential is taxonomically coherent at higher taxonomic ranks. The quantitative analyses presented here characterize the integral relationship between diversification of microbial lineages and the metabolisms which they host.
Oceanic oil-degrading bacteria produce copious amounts of exopolymeric substances (EPS) that facilitate their access to oil. The fate of EPS in the water column is in part determined by activities of heterotrophic microbes capable of utilizing EPS compounds as carbon and energy sources. To evaluate the potential of natural microbial communities to degrade EPS produced during oil degradation, we measured potential hydrolysis rates of six structurally distinct polysaccharides in two roller bottle experiments, using water from a natural oil seep in the northern Gulf of Mexico. The suite of polysaccharides used to measure the initial step in carbon degradation is indicative of polymers within microbial EPS. The treatments included (i) unamended surface or deep waters (whole water), and water amended with (ii) a water-accommodated fraction of oil (WAF), (iii) oil dispersant Corexit 9500, and (iv) WAF chemically-enhanced with Corexit (CEWAF). The oil and Corexit treatments were employed to simulate conditions during the Deepwater Horizon oil spill. Polysaccharide hydrolysis rates in the surface-water treatments were lowest in the WAF treatment, despite elevated levels of EPS in the form of transparent exopolymer particles (TEP). In contrast, the three deep-water treatments (WAF, Corexit, CEWAF) showed enhanced hydrolysis rates and TEP levels (WAF) compared to the whole water. We also observed variations in the spectrum of polysaccharide-hydrolyzing enzyme activities among the treatments. These substrate specificities were likely driven by activities of oil-degrading bacteria, shaping the pool of EPS and TEP as well as degradation products of hydrocarbons and Corexit compounds. A model calculation of potential turnover rates of organic carbon within the TEP pool suggests extended residence times of TEP in oil-contaminated waters, making them prone to serve as the sticky matrix for oily aggregates known as marine oil snow.
We applied theoretical and simulation-based approaches to characterize how microbial community structure influences the amount of sequencing effort to reconstruct metagenomes that are assembled from short-read sequences. First, a coupon collector equation was proposed as an analytical model for predicting sequencing effort as a function of microbial community structure. Characterization was performed by varying community structure properties such as richness, evenness, and genome size. Simulations demonstrated that while community richness and evenness influenced the sequencing effort required to sequence a community metagenome to exhaustion, the effort necessary to sequence an individual genome to a target fraction of exhaustion depended only on the relative abundance of the ge- nome and its genome size. A second analysis evaluated the quantity, completion, and contamination of complete-metagenome-assembled genome equivalents, a bioinformatic pipeline normalized metric for metagenome-assembled genome (MAG) quantity, as a function of sequencing effort on four preexisting sequence read data sets from different environments. These data sets were subsampled to various degrees of completeness to simulate the effect of sequencing effort on MAG retrieval. Modeling suggested that sequencing efforts beyond what is typical in published experiments (1 to 10 Gbp) would generate diminishing returns in terms of MAG bin- ning. A software tool, Genome Relative Abundance to Sequencing Effort (GRASE), was created to assist investigators to further explore this relationship. Reevaluation of the relationship between sequencing effort and binning success in the context of genome relative abundance, as opposed to base pairs, provides a constraint on sequencing experiments based on the relative abundance of microbes in an environ- ment rather than arbitrary levels of sequencing effort.
A recent paper by Martiny argues that “high proportions” of bacteria in diverse Earth environments have been cultured. Here we reanalyze a portion of the data in that paper, and argue that the conclusion is based on several technical errors, most notably a calculation of sequence similarity that does not account for sequence gaps, and the reliance on 16S rRNA gene amplicons that are known to be biased towards cultured organisms. We further argue that the paper is also based on a conceptual error: namely, that sequence similarity cannot be used to infer “culturability” because one cannot infer physiology from 16S rRNA gene sequences. Combined with other recent, more reliable studies, the evidence supports the conclusion that most bacterial and archaeal taxa remain uncultured.
Anoxic subsurface sediments contain communities of heterotrophic microorganisms that metabolize organic carbon at extraordinarily slow rates. In order to assess the mechanisms by which subsurface microorganisms access detrital sedimentary organic matter, we measured kinetics of a range of extracellular peptidases in anoxic sediments of the White Oak River estuary, NC. Nine distinct peptidase substrates were enzymatically hydrolyzed at all depths. Potential peptidase activities (Vmax) decreased with increasing sediment depth, although Vmax expressed on a per cell basis was approximately the same at all depths. Half-saturation constants (Km) decreased with depth, indicating peptidases that functioned more efficiently at low substrate concentrations. Potential activities of extracellular peptidases acting on molecules that are enriched in degraded organic matter (D-phenylalanine and L-ornithine) increased relative to enzymes that act on L-phenylalanine, further suggesting microbial community adaptation to access degraded organic matter. Nineteen classes of predicted, exported peptidases were identified in genomic data from the same site, of which genes for class C25 (gingipain-like) peptidases represented more than 40% at each depth. Methionine aminopeptidases, zinc carboxypeptidases, and class S24-like peptidases, which are involved in single-stranded DNA repair, were also abundant. These results suggest a subsurface heterotrophic microbial community that primarily accesses low-quality detrital organic matter via a diverse suite of well-adapted extracellular enzymes.
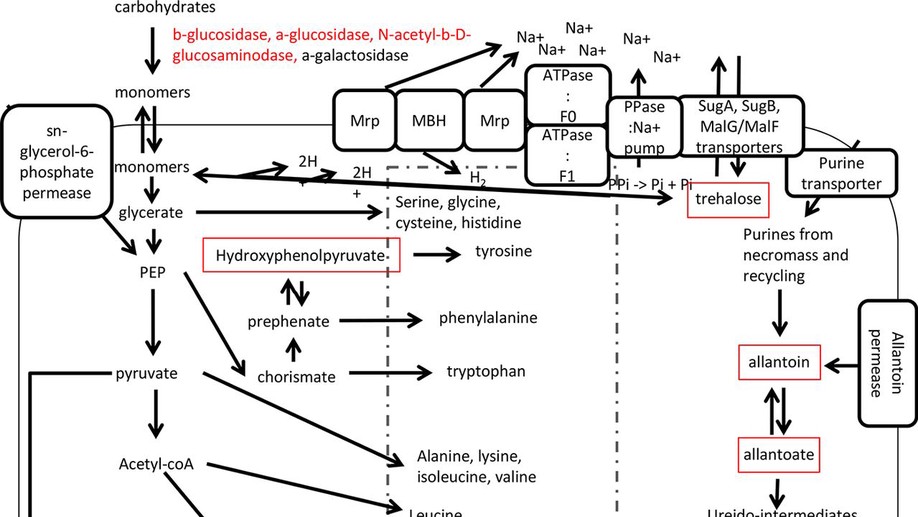
Energy-starved microbes in deep marine sediments subsist at near-zero growth for thousands of years, yet the mechanisms for their subsistence are unknown because no model strains have been cultivated from most of these groups. We investigated Baltic Sea sediments with single-cell genomics, metabolomics, metatranscriptomics, and enzyme assays to identify possible subsistence mechanisms employed by uncultured Atribacteria, Aminicenantes, Actinobacteria group OPB41, Aerophobetes, Chloroflexi, Deltaproteobacteria, Desulfatiglans, Bathyarchaeota, and Euryarchaeota marine group II lineages. Some functions appeared to be shared by multiple lineages, such as trehalose production and NAD+-consuming deacetylation, both of which have been shown to increase cellular life spans in other organisms by stabilizing proteins and nucleic acids, respectively. Other possible subsistence mechanisms differed between lineages, possibly providing them different physiological niches. Enzyme assays and transcripts suggested that Atribacteria and Actinobacteria group OPB41 catabolized sugars, whereas Aminicenantes and Atribacteria catabolized peptides. Metabolite and transcript data suggested that Atribacteria utilized allantoin, possibly as an energetic substrate or chemical protectant, and also possessed energy-efficient sodium pumps. Atribacteria single-cell amplified genomes (SAGs) recruited transcripts for full pathways for the production of all 20 canonical amino acids, and the gene for amino acid exporter YddG was one of their most highly transcribed genes, suggesting that they may benefit from metabolic interdependence with other cells. Subsistence of uncultured phyla in deep subsurface sediments may occur through shared strategies of using chemical protectants for biomolecular stabilization, but also by differentiating into physiological niches and metabolic interdependencies.
Geochemical models typically represent organic matter (OM) as consisting of multiple, independent pools of compounds, each accessed by microorganisms at different rates. However, recent findings indicate that organic compounds can interact within microbial metabolisms. The relevance of interactive effects within marine systems is debated and a mechanistic understanding of its complexities, including microbe-substrate relationships, is lacking. As a first step toward uncovering mediating processes, the interactive effects of distinct pools of OM on the growth and respiration of marine bacteria, individual strains and a simple, constructed community of Roseobacter lineage members were tested. Isolates were provided with natural organic matter (NOM) and different concentrations (1, 4, 40, 400 μM-C) and forms of labile OM (acetate, casamino acids, tryptone, coumarate). The microbial response to the mixed substrate regimes was assessed using viable counts and respiration in two separate experiments. Two marine bacteria and a six-member constructed community were assayed with these experiments. Both synergistic and antagonistic growth responses were evident for all strains, but all were transient. The specific substrate conditions promoting a response, and the direction of that response, varied amongst species. These findings indicate that the substrate conditions that result in OM interactive effects are both transient and species-specific and thus influenced by both the composition and metabolic potential of a microbial community.
This study offers insight into the roles anodic and cathodic processes play in electrochemically activated persulfate (EAP) and screens EAP as a viable technique for ciprofloxacin degradation in wastewater. Sulfate radical formation at a boron-doped diamond (BDD) anode and persulfate activation at a graphite cathode were experimentally elucidated using different electrolytes and electrochemical setups. Rapid ciprofloxacin transformation occurred via pseudo-first-order mechanisms with respect to ciprofloxacin in persulfate electrolyte, reaching 84% removal in 120 min using EAP. Transformation pathways were compared to those in nitrate and sulfate electrolytes. Ciprofloxacin removal rates in the electrochemical system were 88% and 33% faster in persulfate than nitrate and sulfate electrolytes, respectively. Total organic carbon removal rates were 93% and 48% faster in persulfate than nitrate and sulfate, respectively. Use of sulfate electrolyte resulted in removal rates 6–7 times faster than those in nitrate solution. Accelerated removal in sulfate was attributed to anodic sulfate radical formation, while enhanced removal in persulfate was associated with cathodic persulfate activation and nonradical persulfate activation at the BDD anode. Quenching experiments indicated both sulfate radicals and hydroxyl radicals contributed to degradation. Comparisons between platinum and graphite cathodes showed similar cathodic persulfate activation and ciprofloxacin degradation.
Proteins constitute a particularly bioavailable subset of organic carbon and nitrogen in aquatic environments but must be hydrolyzed by extracellular enzymes prior to being metabolized by microorganisms. Activities of extracellular peptidases (protein-degrading enzymes) have frequently been assayed in freshwater systems, but such studies have been limited to substrates for a single enzyme [leucyl aminopeptidase (Leu-AP)] out of more than 300 biochemically recognized peptidases. Here, we report kinetic measurements of extracellular hydrolysis of five substrates in 28 freshwater bodies in the Delaware Water Gap National Recreation Area in the Pocono Mountains (PA, United States) and near Knoxville (TN, United States), between 2013 and 2016. The assays putatively test for four aminopeptidases (arginyl aminopeptidase, glyclyl aminopeptidase, Leu-AP, and pyroglutamyl aminopeptidase), which cleave N-terminal amino acids from proteins, and trypsin, an endopeptidase, which cleaves proteins mid-chain. Aminopeptidase and the trypsin-like activity were observed in all water bodies, indicating that a diverse set of peptidases is typical in freshwater. However, ratios of peptidase activities were variable among sites: aminopeptidases dominated at some sites and trypsin-like activity at others. At a given site, the ratios remained fairly consistent over time, indicating that they are driven by ecological factors. Studies in which only Leu-AP activity is measured may underestimate the total peptidolytic capacity of an environment, due to the variable contribution of endopeptidases.
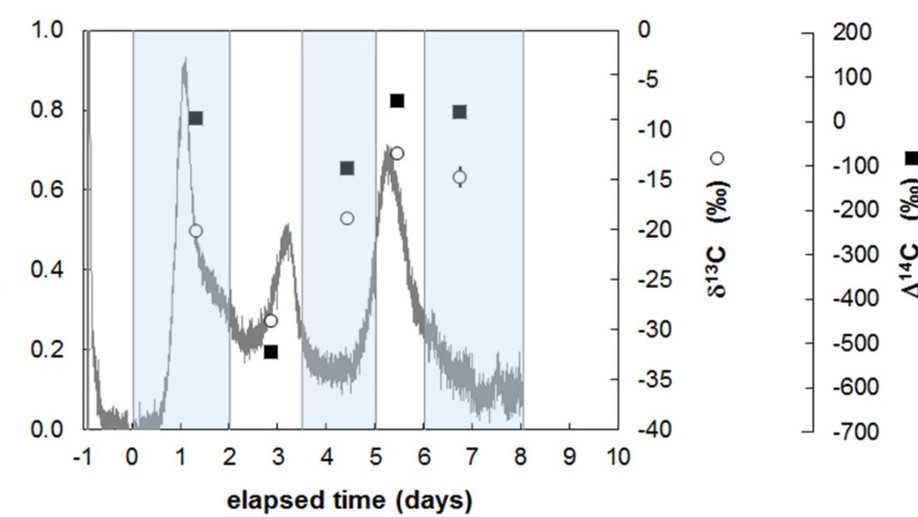
Aquatic sediments harbour diverse microbial communities that mediate organic matter degradation and influence biogeochemical cycles. The pool of bioavailable carbon continuously changes as a result of abiotic processes and microbial activity. It remains unclear how microbial communities respond to heterogeneous organic matrices and how this ultimately affects heterotrophic respiration. To explore the relationships between the degradation of mixed carbon substrates and microbial activity, we incubated batches of organic‐rich sediments in a novel bioreactor (IsoCaRB) that permitted continuous observations of CO2 production rates, as well as sequential sampling of isotopic signatures (δ13C, Δ14C), microbial community structure and diversity, and extracellular enzyme activity. Our results indicated that lower molecular weight (MW), labile, phytoplankton‐derived compounds were degraded first, followed by petroleum‐derived exogenous pollutants, and finally by higher MW polymeric plant material. This shift in utilization coincided with a community succession and increased extracellular enzyme activities. Thus, sequential utilization of different carbon pools induced changes at both the community and cellular level, shifting community composition, enzyme activity, respiration rates, and residual organic matter reactivity. Our results provide novel insight into the accessibility of sedimentary organic matter and demonstrate how bioavailability of natural organic substrates may affect the function and composition of heterotrophic bacterial populations.
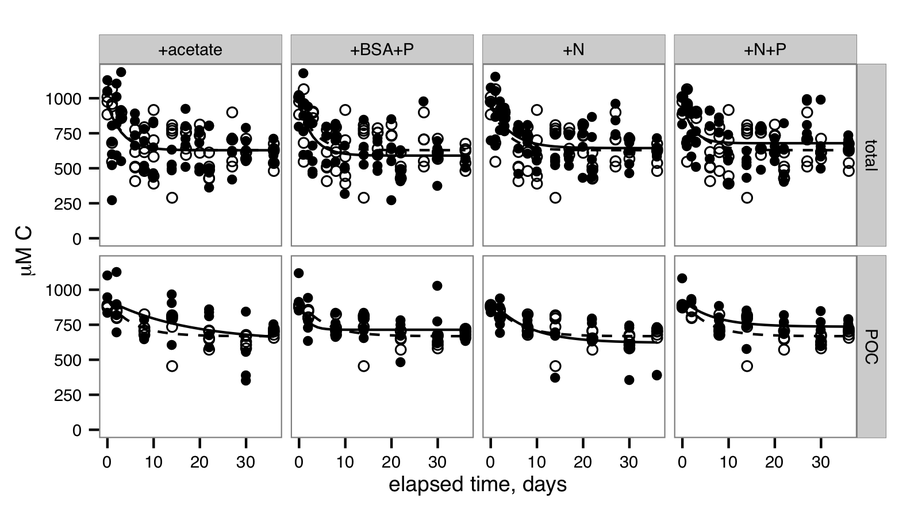
The “priming effect,” in which addition of labile substances changes the remineralization rate of recalcitrant organic matter, has been intensively studied in soils, but is less well-documented in aquatic systems. We investigated the extent to which additions of nutrients or labile organic carbon could influence remineralization rates of 14C-labeled, microbially-degraded, phytoplankton-derived organic matter (OM) in microcosms inoculated with microbial communities drawn from Grove Creek Estuary in coastal Georgia, USA. We found that amendment with labile protein plus phosphorus increased remineralization rates of degraded, phytoplankton-derived OM by up to 100%, whereas acetate slightly decreased remineralization rates relative to an unamended control. Addition of ammonium and phosphate induced a smaller effect, whereas addition of ammonium alone had no effect. Counterintuitively, alkaline phosphatase activities increased in response to the addition of protein under P-replete conditions, indicating that production of enzymes unrelated to the labile priming compound may be a mechanism for the priming effect. The observed priming effect was transient: after 36 days of incubation roughly the same quantity of organic carbon had been mineralized in all treatments including no-addition controls. This timescale is on the order of the typical hydrologic residence times of well-flushed estuaries suggesting that priming in estuaries has the potential to influence whether OC is remineralized in situ or exported to the coastal ocean.
The identities and biochemical properties of extracellular enzymes present in natural environments are poorly constrained. We used a series of competitive inhibition experiments with samples from a freshwater environment (the Tennessee River at Knoxville, TN, USA) and a marine environment (Bogue Sound, NC, USA) to characterize the range of substrate specificities of naturally occurring enzymes that hydrolyze L-leucine 7-amido-4-methylcoumarin (Leu-AMC), L‑proline-AMC (Pro-AMC), and L-arginine-AMC (Arg-AMC)—putative substrates for leucyl-aminopeptidase, prolyl-aminopeptidase, and arginyl-aminopeptidase, respectively. Extracellular peptidases which hydrolyzed Arg-AMC and Leu-AMC demonstrated affinity for up to 8 other amino acids, whereas those hydrolyzing Pro-AMC in the Tennessee River, and Arg-AMC at Bogue Sound, were more specific to proline and arginine, respectively. Patterns of substrate affinity showed that Leu-AMC (at both sampling sites) and Arg-AMC (at Bogue Sound) were primarily hydrolyzed by enzymes other than leucyl-aminopeptidase and arginyl-aminopeptidase, respectively. The set of naturally occurring peptidases in both environments showed greater affinity towards a subset of amino acids. These amino acids were on average larger, yielded more free energy from oxidation to CO2, and tended to be depleted in aged organic matter. These relationships indicate that pathways of amino acid diagenesis are at least partially controlled by the substrate specificities of the peptidases involved in protein degradation.
Marine sediments host a large population of diverse, heterotrophic, uncultured microorganisms with unknown physiologies that control carbon flow through organic matter decomposition. Recently, single-cell genomics uncovered new key players in these processes, such as the miscellaneous crenarchaeotal group. These widespread archaea encode putative intra- and extracellular proteases for the degradation of detrital proteins present in sediments. Here, we show that one of these enzymes is a self-compartmentalizing tetrameric aminopeptidase with a preference for cysteine and hydrophobic residues at the N terminus of the hydrolyzed peptide. The ability to perform detailed characterizations of enzymes from native subsurface microorganisms, without requiring that those organisms first be grown in pure culture, holds great promise for understanding key carbon transformations in the environment as well as identifying new enzymes for biomedical and biotechnological applications.
Extracellular enzymes produced by heterotrophic microbial communities are major drivers of carbon and nutrient cycling in terrestrial, freshwater, and marine environments. Although carbon and nutrient cycles are coupled on global scales, studies of extracellular enzymes associated with terrestrial, freshwater, and marine microbial communities are not often compared across ecosystems. In part, this disconnect arises because the environmental parameters that control enzyme activities in terrestrial and freshwater systems, such as temperature, pH, and moisture content, have little explanatory power for patterns of enzyme activities in marine systems. Instead, factors such as the functional diversity of microbial communities may explain varying patterns of enzyme activities observed in the ocean to date. In any case, many studies across systems focus on similar issues that highlight the commonalities of microbial community organization. Examples include the effective lifetime of enzymes released into the environment; the extent to which microbial communities coordinate enzyme expression to decompose complex organic substrates; and the influence of microbial community composition on enzyme activities and kinetics. Here we review the often-disparate research foci in terrestrial, freshwater, and marine environments. We consider the extent to which environmental factors may regulate extracellular enzyme activities within each ecosystem, and highlight commonalities and current methodological challenges to identify research questions that may aid in integrating crosssystem perspectives in the future.
In Arctic marine bacterial communities, members of the phylum Verrucomicrobia are consistently detected, although not typically abundant, in 16S rRNA gene clone libraries and pyrotag surveys of the marine water column and in sediments. In an Arctic fjord (Smeerenburgfjord) of Svalbard, members of the Verrucomicrobia, together with Flavobacteria and smaller proportions of Alpha- and Gammaproteobacteria, constituted the most frequently detected bacterioplankton community members in 16S rRNA gene-based clone library analyses of the water column. Parallel measurements in the water column of the activities of six endo-acting polysaccharide hydrolases showed that chondroitin sulfate, laminarin, and xylan hydrolysis accounted for most of the activity. Several Verrucomicrobia water column phylotypes were affiliated with previously sequenced, glycoside hydrolase-rich genomes of individual Verrucomicrobia cells that bound fluorescently labeled laminarin and xylan and therefore constituted candidates for laminarin and xylan hydrolysis. In sediments, the bacterial community was dominated by different lineages of Verrucomicrobia, Bacteroidetes, and Proteobacteria but also included members of multiple phylum-level lineages not observed in the water column. This community hydrolyzed laminarin, xylan, chondroitin sulfate, and three additional polysaccharide substrates at high rates. Comparisons with data from the same fjord in the previous summer showed that the bacterial community in Smeerenburgfjord changed in composition, most conspicuously in the changing detection frequency of Verrucomicrobia in the water column. Nonetheless, in both years the community hydrolyzed the same polysaccharide substrates.
The identities and biochemical properties of extracellular enzymes present in natural environments are poorly constrained. We used a series of competitive inhibition experiments with samples from a freshwater environment (the Tennessee River at Knoxville, TN, USA) and a marine environment (Bogue Sound, NC, USA) to characterize the range of substrate specificities of naturally occurring enzymes that hydrolyze L-leucine 7-amido-4-methylcoumarin (Leu-AMC), L‑proline-AMC (Pro-AMC), and L-arginine-AMC (Arg-AMC)—putative substrates for leucyl-aminopeptidase, prolyl-aminopeptidase, and arginyl-aminopeptidase, respectively. Extracellular peptidases which hydrolyzed Arg-AMC and Leu-AMC demonstrated affinity for up to 8 other amino acids, whereas those hydrolyzing Pro-AMC in the Tennessee River, and Arg-AMC at Bogue Sound, were more specific to proline and arginine, respectively. Patterns of substrate affinity showed that Leu-AMC (at both sampling sites) and Arg-AMC (at Bogue Sound) were primarily hydrolyzed by enzymes other than leucyl-aminopeptidase and arginyl-aminopeptidase, respectively. The set of naturally occurring peptidases in both environments showed greater affinity towards a subset of amino acids. These amino acids were on average larger, yielded more free energy from oxidation to CO2, and tended to be depleted in aged organic matter. These relationships indicate that pathways of amino acid diagenesis are at least partially controlled by the substrate specificities of the peptidases involved in protein degradation.
To describe a microbe’s physiology, including its metabolism, environmental roles, and growth characteristics, it must be grown in a laboratory culture. Unfortunately, many phylogenetically novel groups have never been cultured, so their physiologies have only been inferred from genomics and environmental characteristics. Although the diversity, or number of different taxonomic groups, of uncultured clades has been studied well, their global abundances, or numbers of cells in any given environment, have not been assessed. We quantified the degree of similarity of 16S rRNA gene sequences from diverse environments in publicly available metagenome and metatranscriptome databases, which we show have far less of the culture bias present in primer-amplified 16S rRNA gene surveys, to those of their nearest cultured relatives. Whether normalized to scaffold read depths or not, the highest abundances of metagenomic 16S rRNA gene sequences belong to phylogenetically novel uncultured groups in seawater, freshwater, terrestrial subsurface, soil, hypersaline environments, marine sediment, hot springs, hydrothermal vents, nonhuman hosts, snow, and bioreactors (22% to 87% uncultured genera to classes and 0% to 64% uncultured phyla). The exceptions were human and human-associated environments, which were dominated by cultured genera (45% to 97%). We estimate that uncultured genera and phyla could comprise 7.3 × 1029 (81%) and 2.2 × 1029 (25%) of microbial cells, respectively. Uncultured phyla were overrepresented in metatranscriptomes relative to metagenomes (46% to 84% of sequences in a given environment), suggesting that they are viable. Therefore, uncultured microbes, often from deeply phylogenetically divergent groups, dominate nonhuman environments on Earth, and their undiscovered physiologies may matter for Earth systems.
The assembly of single-amplified genomes (SAGs) and metagenome-assembled genomes (MAGs) has led to a surge in genome-based discoveries of members affiliated with Archaea and Bacteria, bringing with it a need to develop guidelines for nomenclature of uncultivated microorganisms. The International Code of Nomenclature of Prokaryotes (ICNP) only recognizes cultures as ‘type material’, thereby preventing the naming of uncultivated organisms. In this Consensus Statement, we propose two potential paths to solve this nomenclatural conundrum. One option is the adoption of previously proposed modifications to the ICNP to recognize DNA sequences as acceptable type material; the other option creates a nomenclatural code for uncultivated Archaea and Bacteria that could eventually be merged with the ICNP in the future. Regardless of the path taken, we believe that action is needed now within the scientific community to develop consistent rules for nomenclature of uncultivated taxa in order to provide clarity and stability, and to effectively communicate microbial diversity.